
This Evolution 101 post is by MSU grad student Tyler Derr
One of the basic mechanisms of evolutionary change is natural selection. It was in Charles Darwin’s famous book, On the Origin of Species, where he defined natural selection to be the “principle by which each slight variation [of a trait], if useful, is preserved.” (Darwin 1859). Knowing that many people would be skeptical of what he presented in his book, the first chapter is structured to first discuss selection in terms of breeding. He presented examples of where humans have selectively bred both animals and plants.
Artificial selection (also called selective breeding) is a process in which humans interfere with natural selection to obtain certain traits we desire an organism to have. This process is performed by choosing which animals or plants are allowed to mate with each other in the hopes if both of the parents have a certain observable trait then their offspring will as well. Shown in Fig. 1 are common vegetables that have all been cultivated from wild mustard by past farmers artificially selecting traits in the plant.

Fig. 1. Vegetables that arose from the wild mustard due to selecting for different traits.
Selective breeding has been done on many animals. Examples of such artificial selection being performed can be seen in dogs. Due to this selective breeding, there are now hundreds of different breeds. It was only just recently, thanks to whole genome sequencing, that we have discovered that gray wolves and dogs started to diverge from a common ancestor at the same time roughly 27,000- 40,000 years ago (Skoglund 2015). Although today we breed dogs for certain traits such as cute floppy ears or a fluffy coat, we can pretty safely say that these would have probably not been at the top of the list with humans that long ago. We know that based on when dogs first arose this would have been the time when humans were still hunter-gathers (Freedman 2014). Fig. 2 below shows a few dog breeds representing were they were selectively breed, based on geographical location, and also representing from what previous dog breeds they were breed from.
So if humans tens of thousands of years ago did not select for all the same traits that we select for today, what did they desire? Well, it can be hypothesized that they simply desired the dogs to be non-aggressive. Although we normally think of evolution as a very long process (which it is with natural selection), the game changes when we as humans start getting involved. It was in 1959 that a Russian geneticist Kmitry K. Belyaev (shown in Fig. 3) began a study in the hopes of breeding a population of tame foxes (Trut 1999). Belyaev solely selected for tameness and strictly against aggression when breeding. After just ten generations, 18% of the pups were not only tame, but showing signs of affection such as whimpering for attention and even licking the experimenters (Trut 1999). Evolution in Action: The Silver Fox Experiment, is a short clip from the BBC’s documentary [The Secret Life of Dogs] in which they discuss the early stages of the fox experiment and then also show the progress that has been made over the last 50 years.
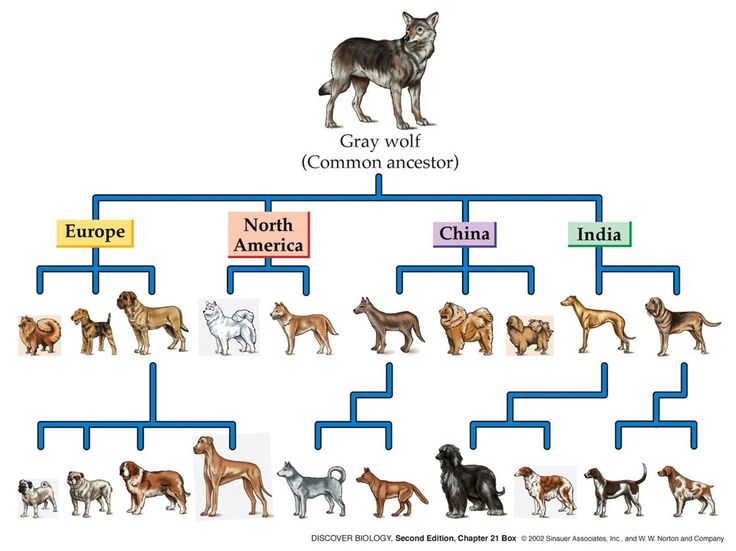
Fig. 2: Examples of how selective dog breeding has branched from the common ancestor based on geographical location. Cain, Michael L., Damman, Hans, Lue, Robert A. and Carol Kaesuk Yoon. Discover Biology Second Edition. New York: W. W. Norton, 2002.
As mentioned before, the only trait that was selected for in the fox experiment was tameness, however interestingly enough quite a number of other traits came with it. Other physical traits such as a curly tail instead of straight, floppy ears, and shorter limbs began to appear in the tame foxes, which are traits commonly shared among other domesticated animals (Trut 1999). What was just described is commonly known as the correlation of traits, which is another interesting topic to mention when discussing artificial selection. This is when selecting for a specific trait not only allows the offspring to have the trait selected for, but also a set of other traits that are genetically correlated.

Fig. 3: Belyaev shown with some of the tame foxes that were bred in his experiment.
As seen from both the plant and animal examples, humans can have a large impact on a species when performing selective breeding. It will be interesting to see how future organisms will be changed to fit the wants and or needs of humans in the future.
References:
Darwin, C. (1859). “On the origin of species by means of natural selection, or by preservation of favoured races in the struggle for life”. London: John Murray.
Freedman, A., et al. (2014). “Genome sequencing highlights the dynamic early history of dogs”. PLoS genetics 10(1): e1004016.
Skoglund, P., et al. (2015). “Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds”. Current Biology 25(11): 1515–9.
Trut, L. (1999). “Early Canid Domestication: The Farm-Fox Experiment Foxes bred for tamability in a 40-year experiment exhibit remarkable transformations that suggest an interplay between behavioral genetics and development”. American Scientist 87(2): 160-169.







 Greetings! My name is Siobahn Day. I’m currently a PhD student in the Computer Science at North Carolina A&T State University. I work as a graduate researcher in the Center for Advanced Studies in Identity Science. I have developed the concept of Adversarial Authorship as a means of preserving author anonymity. I’m currently developing and evaluating an Interactive Evolutionary Computation for Adversarial Authorship which allows users to conceal their writing style.
Greetings! My name is Siobahn Day. I’m currently a PhD student in the Computer Science at North Carolina A&T State University. I work as a graduate researcher in the Center for Advanced Studies in Identity Science. I have developed the concept of Adversarial Authorship as a means of preserving author anonymity. I’m currently developing and evaluating an Interactive Evolutionary Computation for Adversarial Authorship which allows users to conceal their writing style.





{kind=link}