This week’s BEACON Researchers at Work blog post is by University of Texas at Austin Research Scientist Dhivya Arasappan.

Bioinformatics is an interdisciplinary field in which computer algorithms and statistical methods are applied to answer biological questions. It is a field that is often talked about as the next biggest thing; it is a field I knew nothing about when I started college. As it happens with many things in life, I never planned on becoming a bioinformatician, but I’m happy that I did. After completing my undergraduate studies in Computer Science, I was underwhelmed by the standard career opportunities that were available to me. I was interested in applying computer algorithms towards a greater goal. This is when I learned about the field of bioinformatics and I chose to to do my graduate work in it. Now after 7 years as a bioinformatician, I can say that the challenges in the field are more interesting and the demand greater than ever. With the advent of next generation sequencing, our ability to generate biological data is rapidly outpacing our ability to store and make sense of it. This has made bioinformatics crucial both in research and industry settings.
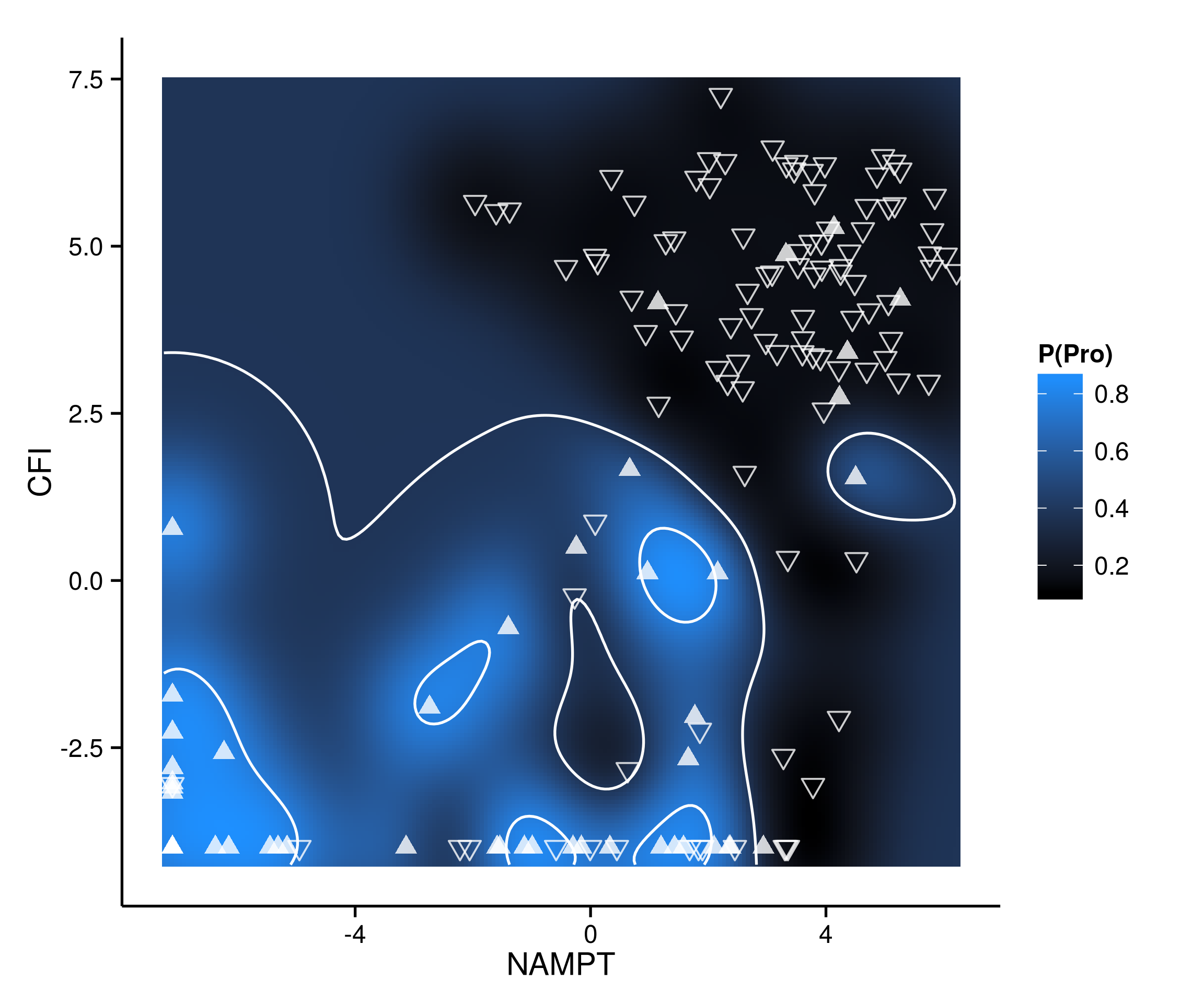
 At the University of Texas at Austin, I work at the Center for Computational Biology and Bioinformatics (CCBB) as part of the bioinformatics consulting group. At its core, what I do as a bioinformatician is parse through large amounts of data to identify patterns that may be biologically meaningful. A large and often most exciting aspect of my job is collaborating with labs to guide experimental design and perform computational analysis of their high throughput data sets. For the last two years, I’ve worked with Dr. Bob Jansen’s lab to sequence, assemble and annotate a medicinally important desert plant known as Rhazya stricta. This plant grows abundantly in arid environments in the Middle East and India and belongs to the Apocynaceae family and Gentianales order. Like others in the Gentianales order, Rhazya is a producer of monoterpenoid indole alkaloids. These compounds are of great interest because several have antibacterial activity and some have found use as anticancer agents. We assembled and annotated the nuclear genome of Rhazya stricta in order to better understand the pathways related to the generation of these compounds. For the de novo assembly of the genome, we generated data from multiple sequencing platforms. Each sequencing platform comes with inherent strengths and weaknesses. Some, like PacBio, produce very long reads which are conducive to whole genome assembly, but are low in yield and high in error. Other platforms, like the Illumina HiSeq, produce a high yield of high quality reads, but the reads are short in length. By using a complementary set of data from multiple platforms, we were able to generate a high quality genome assembly. Bioinformatically, it is a challenge to find a genome assembler that is well equipped to handle data from multiple platforms. These challenges are compounded by the fact that each platform has different error rates and is prone to different types of sequencing errors. We used an iterative assembly method by pipelining multiple assembly, gap filling and scaffolding tools in sequence to generate a high quality draft genome in a reasonable amount of time. By annotating this genome, we’ve been able to elucidate some of the metabolic pathways in the plant.
At the University of Texas at Austin, I work at the Center for Computational Biology and Bioinformatics (CCBB) as part of the bioinformatics consulting group. At its core, what I do as a bioinformatician is parse through large amounts of data to identify patterns that may be biologically meaningful. A large and often most exciting aspect of my job is collaborating with labs to guide experimental design and perform computational analysis of their high throughput data sets. For the last two years, I’ve worked with Dr. Bob Jansen’s lab to sequence, assemble and annotate a medicinally important desert plant known as Rhazya stricta. This plant grows abundantly in arid environments in the Middle East and India and belongs to the Apocynaceae family and Gentianales order. Like others in the Gentianales order, Rhazya is a producer of monoterpenoid indole alkaloids. These compounds are of great interest because several have antibacterial activity and some have found use as anticancer agents. We assembled and annotated the nuclear genome of Rhazya stricta in order to better understand the pathways related to the generation of these compounds. For the de novo assembly of the genome, we generated data from multiple sequencing platforms. Each sequencing platform comes with inherent strengths and weaknesses. Some, like PacBio, produce very long reads which are conducive to whole genome assembly, but are low in yield and high in error. Other platforms, like the Illumina HiSeq, produce a high yield of high quality reads, but the reads are short in length. By using a complementary set of data from multiple platforms, we were able to generate a high quality genome assembly. Bioinformatically, it is a challenge to find a genome assembler that is well equipped to handle data from multiple platforms. These challenges are compounded by the fact that each platform has different error rates and is prone to different types of sequencing errors. We used an iterative assembly method by pipelining multiple assembly, gap filling and scaffolding tools in sequence to generate a high quality draft genome in a reasonable amount of time. By annotating this genome, we’ve been able to elucidate some of the metabolic pathways in the plant.
 Along with our collaboration and research efforts, another important facet of the consulting group is training. We provide numerous educational opportunities for researchers from within UT and outside to learn bioinformatics skills. These skills become especially vital when the researchers are bombarded with their own large-scale data sets and need to parse something meaningful out of them. I have had an opportunity to teach and train many graduate students, post-docs and professors in the last 2 years and it has been a very rewarding experience. As part of our Big Data in Biology Summer School program, I teach an Introduction to RNA-Seq course that allows students to get hands-on skills in analyzing RNA-Seq data sets. Apart from this longer course, I also teach 3-hour short courses during the fall and spring semesters on topics related to data analysis. We also strive to develop the bioinformatics community within the University and on that front, I run a monthly meeting of bioinformaticians called byte club. A play on the movie title Fight Club, byte club, offers a place for people doing bioinformatics and people interested in bioinformatics to listen to an interesting talk, communicate with each other and hopefully resolve issues that they may be facing.
Along with our collaboration and research efforts, another important facet of the consulting group is training. We provide numerous educational opportunities for researchers from within UT and outside to learn bioinformatics skills. These skills become especially vital when the researchers are bombarded with their own large-scale data sets and need to parse something meaningful out of them. I have had an opportunity to teach and train many graduate students, post-docs and professors in the last 2 years and it has been a very rewarding experience. As part of our Big Data in Biology Summer School program, I teach an Introduction to RNA-Seq course that allows students to get hands-on skills in analyzing RNA-Seq data sets. Apart from this longer course, I also teach 3-hour short courses during the fall and spring semesters on topics related to data analysis. We also strive to develop the bioinformatics community within the University and on that front, I run a monthly meeting of bioinformaticians called byte club. A play on the movie title Fight Club, byte club, offers a place for people doing bioinformatics and people interested in bioinformatics to listen to an interesting talk, communicate with each other and hopefully resolve issues that they may be facing.
 Another exciting avenue for bioinformatics training that is opening up at UT is a new stream as part of the freshman research initiative called ‘Big data in Biology’. The Freshman Research Initiative (FRI) provides first-year students the opportunity to participate in real research with UT faculty and staff. It has been a very successful program for the last 10 years and this spring, FRI is introducing three new technology streams that focus specifically on improving undergraduates’ industry relevant technological skills. I will be the technology educator responsible for the Big Data in Biology stream and I’m very excited about the prospect of designing a curriculum and research projects to impart undergraduates with skills in large-scale data analysis.
Another exciting avenue for bioinformatics training that is opening up at UT is a new stream as part of the freshman research initiative called ‘Big data in Biology’. The Freshman Research Initiative (FRI) provides first-year students the opportunity to participate in real research with UT faculty and staff. It has been a very successful program for the last 10 years and this spring, FRI is introducing three new technology streams that focus specifically on improving undergraduates’ industry relevant technological skills. I will be the technology educator responsible for the Big Data in Biology stream and I’m very excited about the prospect of designing a curriculum and research projects to impart undergraduates with skills in large-scale data analysis.
As a member of the bioinformatics consulting group, I believe I help enable cutting-edge research, both by collaborating with labs on all aspects of their projects and by educating the community on bioinformatics skills. This is very fulfilling and it makes going to work every day a joy.
For more information about Dhivya’s work, you can contact her at darasappan at mail dot utexas dot edu.