
This week’s BEACON Researchers at Work blog post is by MSU graduate student Gaurav Moghe.
 There are an estimated 9 million eukaryotic species on our planet, of which only 1.2 million (~15%) have been catalogued so far. Of these 1.2 million, only a few dozen are used as model organisms in modern science. In other words, most of the biological knowledge that mankind has, is derived from <0.001% of species!
There are an estimated 9 million eukaryotic species on our planet, of which only 1.2 million (~15%) have been catalogued so far. Of these 1.2 million, only a few dozen are used as model organisms in modern science. In other words, most of the biological knowledge that mankind has, is derived from <0.001% of species!
This is shocking, but there is a good reason for that. The model organisms are assumed to be representative of their taxa, a fair assumption given common descent. Saccharomyces cerevisiae is representative of fungi, Drosophila melanogaster is representative of invertebrates and Mus musculus is representative of mammals. Common descent also ensures that some of the principles learnt in yeasts are also applicable to mammals and vice-versa. This strategy has worked very well so far and has provided us incredible insights into the mechanisms that keep life up and running. However, not all principles learnt in one species can be extrapolated to others. For example, plants belonging to the same genus can look very different and can have variations in their developmental, metabolic and stress response pathways. Even populations of the same species inhabiting different habitats can vary due to local adaptation or drift. A large proportion of the molecular mechanisms and evolutionary pathways responsible for the observed natural variation remain to be discovered in these un-sampled species; however, till very recently this was not possible partly due to technological limitations.

Response of three different Arabidopsis thaliana accessions to 3 weeks of 500mM salt stress. The molecular basis of such variation, how the genes influencing such variation evolved and the effect of such variation on fitness in the “real world” is poorly understood.
Over the past decade, the advent of new technologies has allowed us to explore beyond the model organisms. High-throughput assay technologies such as microarrays, Illumina sequencing and mass spectrometry, coupled with tremendous increases in computing power have allowed us to sample the genomes, transcriptomes and now even proteomes and metabolomes of a variety of organisms. The resultant data explosion has given us the opportunity to not only sample natural variation in the living world but also to understand what causes this variation using comparative “omic” approaches. When I joined the Shiu lab in 2008, I was fascinated by the diversity in the plant world and wanted to learn more about comparative genomics and molecular evolution. My research has made use of recently-available high-throughput data to understand the evolutionary principles associated with certain biological processes.
Studies in the Shiu Lab have focused on three themes – identifying novel genes in genomes, understanding the evolutionary patterns of duplicate genes and understanding how the expression of genes is regulated – using a combination of computational and experimental strategies in plants. In my own research, I partly worked on addressing the nature of intergenic transcription in Arabidopsis thaliana. Spurred by the massive deluge of studies and popular news articles proclaiming the end of “junk DNA” in genomes, we sought to understand whether the claims of pervasive intergenic transcription and functionality of intergenic transcripts hold true if A. thaliana transcription was analyzed using Illumina sequencing data, rather than tiling array data which is of lower quality. We also used comparative genomics to assess conservation of these intergenic transcripts across fifteen sequenced plant genomes and between 80 A. thaliana accessions, whose genomes are now available as part of the 1001 Arabidopsis Genome Project. We found that only 5-10% of the A. thaliana intergenic space is transcribed and that around the same percent shows any signature of selection within or between species, suggesting widespread prevalence of transcriptional noise. Using multiple characteristics of intergenic transcripts such as their expression level, breadth of expression, distance from annotated genes, transcript length and degree of constraint, one can estimate the probability of an intergenic transcript being functional vs noise, thus aiding identification of non-canonical, novel genes in the intergenic space in different plant genomes.
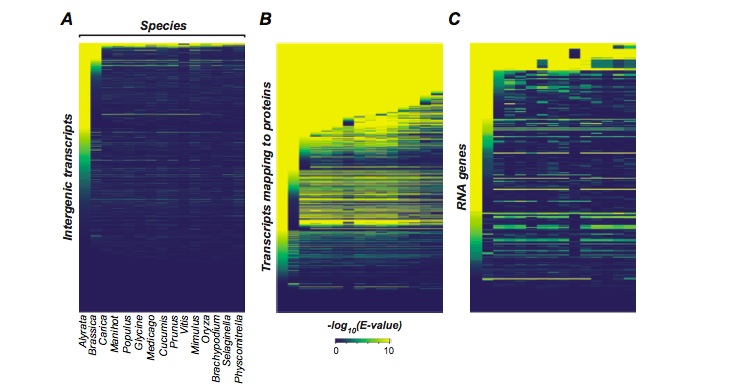
Conservation of intergenic transcripts (A) compared to transcripts mapping to protein-coding genes (B) and RNA genes (C). X-axis represents genomes of different plant species and each row on Y-axis represents an individual feature. The color shades indicate the significance of the BLAST hit of the feature in the corresponding plant genome, with yellow being higher significance. This figure shows that intergenic transcripts are rapidly lost through evolutionary time, at a faster rate than transcripts mapping to protein-coding genes and RNA genes.
My research has also focused on understanding the evolutionary patterns of duplicate genes derived from whole genome duplication (WGD) in the Brassicaceae family of plants. WGD is a ubiquitous phenomenon among flowering plants and duplicate genes produced via WGD may aid in functional diversification and/or adaptation of the polyploid species. As part of my research, we sequenced and annotated the genome of wild radish (Raphanus raphanistrum) and compared the patterns of evolution of duplicate genes and pseudogenes between multiple Brassicaceae species. My results reveal a complex pattern of gene loss and retention post WGD.
The evolutionary biologist Theodosius Dobzhansky famously wrote, “Nothing in biology makes sense except in the light of evolution.” This statement is ever more true today. Today, using high-throughput sequencing methods, we can sequence transcripts being produced at a concentration of 1 transcript/1000 cells. We have the ability to rapidly sample all the metabolites being synthesized in cells and tissues. Population level genome sequencing can reveal thousands to millions of polymorphisms between individuals. However, we still have an unclear idea of the significance of such massive molecular-level variation. How do we filter out truly functional features from random noise? How have such features evolved to their present state? With our newfound ability to sample populations and closely related species, we are beginning to better understand how genes and pathways evolve through time. Molecular evolutionary and comparative genomic approaches are also being used to pinpoint genes responsible for complex phenotypes such as intelligence, identify polymorphisms that may result in disease and define relationships between organisms. Such approaches, coupled with advancements in other technologies, are allowing us to better understand the diversity of the world we live in. It is truly an exciting time to be in evolutionary biology!
For more information about Gaurav’s work, you can contact him at moghegau at msu dot edu.
