
This week’s BEACON Researchers at Work blog post is by University of Texas at Austin research associate Rebecca Young.
Me as a teenager in 1996 (Austin, TX).
From an early age I spent my time outside – chasing lizards, riding horses, and begging to go to a zoo. As this fascination matured I found it was the diversity of life, as Darwin so eloquently describes the “endless forms most beautiful and most wonderful”, which intrigued me most. Where does this variation come from? How are these differences generated during development?
Ironically, while my curiosity and decision to pursue a career in biology arose from a fascination with animal diversity, my study of biology is largely centered on what is shared among organisms. It is now known that the tremendous diversity of animal species on earth develop from remarkably conserved ‘toolkits’ of gene regulatory networks redeployed in context- and species-specific ways. In hindsight, some may argue this is not surprising. The foreleg of a horse and wing of a bat look and function differently, but at their core they are quite similar. Both are appendages, out growths from a remarkably similar bilaterally symmetrical bodyplan; their differences (e.g., their length, width, and arrangement of bones) can be achieved by changes in growth and adjustments in apoptosis during development. However, the underlying developmental and genetic similarities among organisms go beyond homologous traits like limbs – characters that occur in multiple species derived from the same ancestral trait. Developmental genetic similarities appear in non-homologous traits as well. For example, in cases of parallel evolution, where similar features evolve independently in multiple lineages, similar developmental mechanisms can be found (e.g., the electric organ in electric fishes). In some cases traits that have seemingly no similarities evolutionarily or functionally (e.g., human diseases and yeast phenotypes) can share gene regulatory networks (read about ‘phenologs’ here: http://www.phenologs.org/). That traits such as these, having no business being developmentally similar, are in fact “deeply homologous” – i.e., share gene regulatory mechanisms – is a principal discovery resulting from evodevo thinking in biology.

Paired monogamous and non-monogamous species used in our research.
For the majority of its history, the field of evolutionary developmental biology has focused on morphological and physiological characters. However, other types of traits, such as behaviors, should likewise share gene regulatory mechanisms. My work as a research associate in the Hofmann Lab at the University of Texas at Austin (http://cichlid.biosci.utexas.edu/) asks whether similar behaviors, in this case monogamy, that have evolved independently in multiple taxa result from deployment of the same ‘deeply homologous’ gene regulatory mechanism. To answer this question we are taking a comparative transcriptomic approach. Specifically, we quantify expression of genes in the brains of reproductive males in paired, closely-related monogamous and non-monogamous species of voles, mice, song birds, dendrobatid frogs, and cichlid fishes using the next generation sequencing approach RNA-seq. By comparing neural gene expression between monogamous and non-monogamous species within a group (e.g., within voles) we can identify the genes that are up-or down-regulated in the monogamous species. If we do this in all of the groups we can identify the genes that are differentially regulated across all monogamous species examined. This is a jumping off point. From here, we can further examine this list of differentially expressed genes to identify groups of co-expressed genes. We can assess the known interactions and functions of the differentially expressed genes to identify the types of neurogenomic changes that accompany the transitions to monogamy in all of these groups generating functional hypotheses for future experiments.
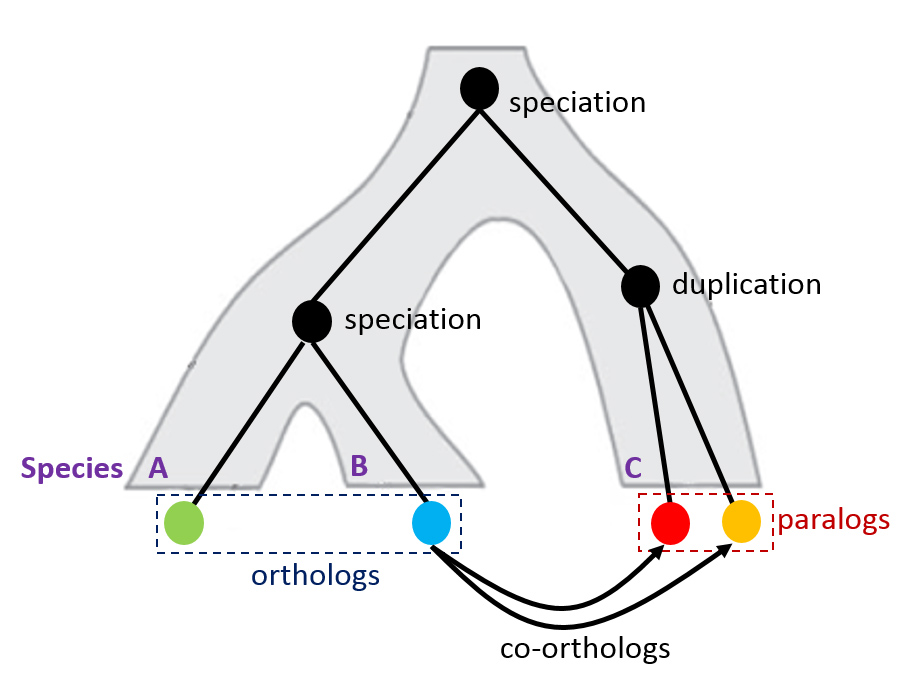
Illustration of orthologs and paralogs.
Getting a list of genes differentially regulated in all these independent evolutionary transitions to monogamy is no small feat. Outside of the efforts required to recruit a consortium of experts who can provide the appropriate tissue for each of these distinct species, comparative approaches in next generation sequencing data analysis are in their infancy. To date, comparative ‘omics research has focused largely on closely related species. When studies do span large evolutionary distances they focus on model systems with well-developed genomic resources (e.g., a well-annotated, published genomes and functionally annotated genes) that facilitate comparisons. Much of our effort has focused on improving these approaches in non-model species like those explored for this project. One of the major challenges is identifying the ‘same’ (orthologous) genes in each species. This problem comes from differences in genome complexity across organisms. For example, gene, gene family, and whole genome duplications have occurred in some lineages and not others, meaning that quite often a gene has more than one copy – called paralogs. How can you compare expression of one gene in one species with two genes in another? To resolve this problem, I have worked in collaboration with the Center for Computational Biology and Bioinformatics (CCBB: http://ccbb.biosci.utexas.edu/) and the Texas Advanced Computing Center (TACC: https://www.tacc.utexas.edu/) to establish an analysis pipeline based on OrthoMCL (http://www.orthomcl.org/) in a high performance computing environment. Rather than focusing on individual genes, we use OrthoMCL to generate ‘orthologous gene groups’ that contain all of the paralogs for a particular gene. These gene groups are generated by comparing sequences within and between species. When gene sequences within species are more similar to each other than to genes in another species, they are grouped as paralogs into these orthologous gene groups. Expression values of the orthologous gene groups can be calculated and compared directly across species. This is not only critical for our research interests, but facilitates comparative ‘omics in general as next generation sequencing approaches are applied to more and more non-model organisms in a diversity of empirical and experimental contexts.

My 2 year old daughter at the Austin Zoo
and Animal Sanctuary .
Right now the scientific pursuits that drive my curiosity are best explored indoors, in a lab or on a computer rather than in the field. Until my research takes me outside again, and from time to time it does (read about my other research directions here http://devoevo.ccbb.utexas.edu/), the observations of the beautiful and wonderful natural variation I have made on horseback as a teenager, in the field as a scientist, and exploring nature with my kids have generated endless questions on the developmental and evolutionary origins of animal diversity.
For more information about Rebecca’s work, you can contact her at youngrl at utexas dot edu.
