
This week’s BEACON Researchers at Work blog post is by North Carolina A&T graduate student Mina Moradi Kordmahalleh.
 The idea of bioinformatics and how an electrical engineer can work on this topic is quite new and interesting to me. In Iran, I received my bachelor and master’s degrees in Electrical Engineering specializing in the control field. At the beginning of my PhD program, I became familiar with a fascinating area of study known as Gene Regulatory Network (GRN) through my investigation of researchers who have backgrounds in the control field and working on bioinformatics. With my background in data mining, control, and bio inspired algorithms, I was motivated to work in GRN.
The idea of bioinformatics and how an electrical engineer can work on this topic is quite new and interesting to me. In Iran, I received my bachelor and master’s degrees in Electrical Engineering specializing in the control field. At the beginning of my PhD program, I became familiar with a fascinating area of study known as Gene Regulatory Network (GRN) through my investigation of researchers who have backgrounds in the control field and working on bioinformatics. With my background in data mining, control, and bio inspired algorithms, I was motivated to work in GRN.
In biological terms, a gene regulatory network is a collection of DNA segments in a cell, which interact with each other and other substances in the cell. Inferences of GRNs are key to understanding the fundamental cellular processes and revealing the underlying relations among genes.
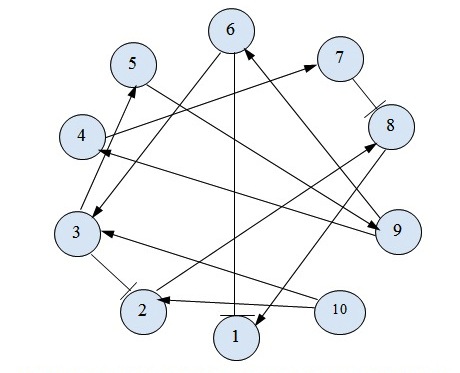
A general representation of a simple gene regulatory network. The nodes in the network represent genes, with the arrow depicting gene activation, and gene inhibition represented with a blunt arrow. Nodes without links indicate no gene interaction.
The ability to model GRNs more accurately may improve medical diagnoses, disease treatment, and drug design. It also helps to understand how drugs affect gene dynamics and which genes are most involved in a process. With the availability of temporal gene expression data in certain biological conditions, different computational methods aim to reconstruct regulatory roles of genes to address biological problems.
The selection of modeling approaches depends on the type and amount of available data, while taking into consideration challenges like high dimensionality, temporal dynamics and measurement noise. The most common mathematical modeling technique for GRNs are ordinary differential equations, Boolean networks, Petri nets, Bayesian networks, Stochastic, and graphical Gaussian models. Different techniques have been proposed to generate models of GRNs to explain a set of time series observations and to predict the behaviors of the components. However, the methods for design and analysis of GRN are in early stages.
In order to clearly show the complexity of the gene network a living cell has been compared to a complex factory, which employs molecular Nano machines while genes and other components of the factory are changing and evolving over time. Factors for these changes include environmental conditions like pH or temperature, maturity stage of an organism where a younger organism may be different from an older one, response to stress like UV irradiation or chemical toxin, and even a disease such as cancer, which all may change the level of protein expression.
The focus of our current work is to address issues such as time lag embedded in the network, external stimuli, and dynamical functioning of the GRN in designing the models. The Artificial Neural Network (ANN) previously was used in constructing the model of GRN because of its ability to handle noisy data and to cover higher order correlations. In this type of network, each node is associated with a particular gene, where the value of the node is the corresponding gene expression. Connections between nodes represent regulatory interaction between genes under a certain weight, which indicates the strength of the connection. Finally, through the training of the ANN, the dynamic of the GRN is constructed based on nonlinear relations among nodes.
Through the effectiveness of ANN in modeling GRN, our current work involves the prediction of the gene expression levels in a gene regulatory network by a new generation of partially connected ANN with evolvable topology. We assume that the observed measurements of genes may depend on some hidden variables. Through the evolutionary process, these hidden variables are firstly able to play the role of memory in order to determine the necessary non-uniform time lag in modeling; and secondly they can play the role of internal states of the biological system such as regulating proteins, excluded genes in the experiments, external signals, and biological noise to represent the latent variables.
Moreover, we assume that partially connected structures exist between nodes, which is consistent with the biological assumption that the genetic networks are usually sparsely connected. Networks can evolve in different ways such as the addition or subtraction of nodes (genes) or parts of the network or by changing the strength of interactions between nodes. In this scheme, the prediction error between actual gene expression data and the estimated ones during the training process guides the search space to discover the network topology with sufficient accuracy on constructing the GRN. Evolution of network topology continues until determining the best fitting model of the GRN, which represents the underlying dynamic behaviors of the genes.
For more information about Mina’s work, you can contact her at mmoradik at aggies dot ncat dot edu.
